
El factor de crecimiento transformante tipo beta (TGF-β) es miembro de una superfamilia de factores polipeptídicos que inhiben el crecimiento e inducen apoptosis en un gran número de tipos celulares, particularmente células epiteliales (1). Se han descrito diferentes clases de receptores de la familia del TGF-β, entre los que destacan el TβRI y el TβRII, que tienen actividad serina-treonina proteína quinasa en su dominio citoplasmático y forman heterodímeros para ser activos. El receptor II tiene una actividad quinasa constitutiva. La unión del TGF-β al receptor II es reconocida por el receptor I, uniéndose a él para formar un complejo. A continuación, el receptor I es fosforilado por el II, lo que estimula su actividad serina-treonina quinasa, con la subsiguiente fosforilación de distintos miembros de la familia de proteínas Smad (R-Smads: Smad2 y Smad3 en el caso del TGF-β1). Estas proteínas fosforiladas se unen a otro miembro de la familia Smad (Co-Smad: Smad 4) y, como resultado, se produce su translocación al núcleo, donde interacciona de forma célula-específica con otros factores de transcripción, para finalmente regular la expresión génica. Una de las principales consecuencias de la unión de TGF-β a sus receptores es su capacidad de inhibir el crecimiento y regular diferenciación y muerte celular. Se ha demostrado que el TGF-β inhibe las actividades de los complejos ciclina D-Cdk4/6 y ciclina E/Cdk2 (reguladores del ciclo celular), lo que conduce a la hipofosforilación de p-Retinoblastoma y a una disminución de la actividad transcripcional de E2Fs. En definitiva, se produce una parada del ciclo celular. Estos efectos inhibidores se han asociado con la capacidad del TGF-β de aumentar la expresión de los inhibidores de ciclinas (CKIs) p15Ink4B, p21Cip1 y p27Kip1. Además, el TGF-β es capaz de inducir muerte celular mediante la producción de especies reactivas de oxígeno y/o activando las rutas de c-Jun-N-terminal quinasa (JNK), o de DAP quinasa (DAPK: Death Associated Protein Kinase), así como interfiriendo con algunas rutas de supervivencia (como la fosfatidilinositol-3-quinasa, PI3-K), activando finalmente la ruta mitocondrial de muerte.
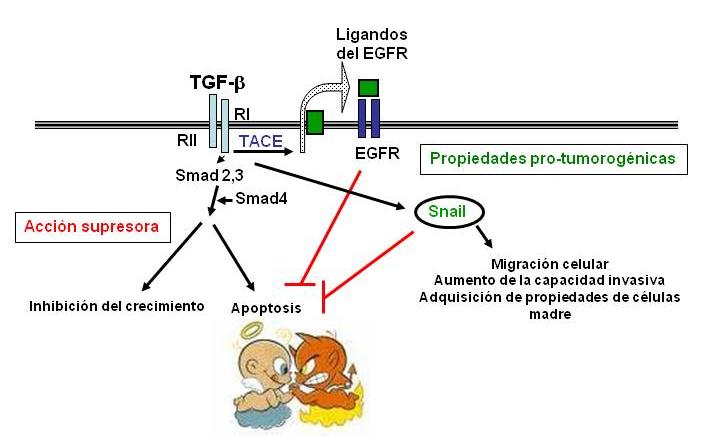
En estadios iniciales de tumorogénesis, un gran número de células pierde su respuesta al TGF-β en términos de inhibición del crecimiento y/o activación de apoptosis (2). Por consiguiente, el TGF-β, sus receptores y las moléculas transductoras son supresores de tumores. Sin embargo, se ha sugerido que la mayoría de los tumores humanos se vuelven refractarios a los efectos inhibidores del crecimiento inducidos por TGF-β debido a des-regulación de otras vías de señalización, por ejemplo, la ruta de Ras/MAPK/ERKs. A modo de ejemplo, la sobreactivación de la vía de las ERKs confiere resistencia a los efectos pro-apoptóticos del TGF-β en células de carcinoma hepatocelular humano, antagonizando su capacidad de inducir la expresión de una proteína esencial para el mecanismo de muerte celular (NOX4) (3). Además, en las células tumorales el TGF-β puede también activar rutas mitogénicas y de supervivencia celular, tales como la ruta de Ras/MAPK y la PI3-K/Akt, bien directamente o mediante la transactivación de otros receptores como el del EGF o el del PDGF (4, 5, ver figura). Un desplazamiento a favor de las rutas pro-mitogénicas y de supervivencia sobre las de inhibición del crecimiento y apoptosis permite a las células escapar a los efectos supresores del TGF-β, manteniendo la respuesta a esta citoquina en otros aspectos, tales como pérdida de expresión de moléculas de adhesión celular, lo que conduce a un aumento de su capacidad migratoria. Además, es conocido que el TGF-β estimula angiogénesis (formación de vasos sanguíneos) y suprime el sistema inmune, procesos que contribuyen a la progresión tumoral (1). Un gran porcentaje de tumores humanos muestran sobre-expresión de TGF-β1, que suele asociarse a mal pronóstico. Durante los últimos años en nuestro laboratorio hemos demostrado que los hepatocitos fetales y las células tumorales del hígado resisten a los efectos apoptóticos del TGF-β y responden a este factor induciendo la expresión del factor de transcripción Snail (6), que media procesos de transición epitelio-mesénquima (TEM) reprimiendo la expresión de genes de adhesión celular, entre los que se encuentra la cadherina-E. Hemos descrito que este proceso también produce des-diferenciación celular (la célula adquiere algunas propiedades de células madre) (7), conduce a un aumento de la capacidad migratoria celular, y, simultáneamente, confiere resistencia a apoptosis (6). En definitiva, las células adquieren un nuevo fenotipo mucho más invasivo y metastático.
Concluyendo, el TGF-β tiene efectos bifásicos durante la tumorogénesis, actuando tempranamente como un supresor tumoral y contribuyendo más tarde a la progresión del tumor, a través de sus acciones paracrinas o autocrinas sobre las células tumorales y su entorno. Las estrategias para el desarrollo de fármacos antitumorales deberán tener en cuenta este comportamiento dual. Es fundamental el avance en los conocimientos de las vías de señalización que originan los diferentes efectos, con el fin de progresar en la búsqueda de drogas que puedan inhibir específicamente sus efectos promotores (movilidad celular, angiogénesis, inmunosupresión, etc.), pero mantengan la respuesta de las células tumorales al TGF-β en términos de inhibición del crecimiento y/o apoptosis.
Referencias:
- Massagué J. TGFβ and cancer. Cell 2008, 134:215-230.
- Seoane J. Escaping from the TGFβ anti-proliferative control. Carcinogenesis 2006, 27:2148-2156.
- Caja L, Sancho P, Bertran E, Iglesias-Serret D, Gil J, Fabregat I. Overactivation of the MEK/ERK pathway in liver tumor cells confers resistance to TGF-{beta}-induced cell death through impairing up-regulation of the NADPH oxidase NOX4. Cancer Res 2009, 69:7595-602.
- Murillo, MM, del Castillo, G, Sánchez, A, Fernández, M and Fabregat, I. Involvement of EGF receptor and c-Src in the survival signals induced by TGF-beta1 in hepatocytes. Oncogene 2005, 24:4580-4587.
- Bruna A, Darken RS, Rojo F, Ocaña A, Peñuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J, Seoane J. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 2007, 11:147-60
- Franco DL, Mainez J, Vega S, Sancho P, Murillo MM, de Frutos CA, Del Castillo G, López-Blau C, Fabregat I, Nieto MA. Snail1 suppresses TGF-beta-induced apoptosis and is sufficient to trigger EMT in hepatocytes. J Cell Sci 2010, 123:3467-77.
- Caja L, Bertran E, Campbell J, Fausto N, Fabregat I. The transforming growth factor-beta (TGF-β) mediates acquisition of a mesenchymal stem cell-like phenotype in human liver cells. J Cell Physiol 2011, 226:1214-23.