
Las hormonas tiroideas, T4 (3,5,3′,5′ tetrayodo-L-tironina, tiroxina) y T3 (3,5,3′ triyodo-L-tironina) son aminoácidos yodados producidos en la glándula tiroides. Regulan muchos procesos del desarrollo y metabolismo de prácticamente todos los tejidos en vertebrados, especialmente el sistema nervioso central. Las enfermedades tiroideas son de las más frecuentes, y las carencias de yodo, así como el hipotiroidismo durante el embarazo y la lactancia pueden afectar seriamente la estructura y función del cerebro de la progenie (cretinismo). La importancia de estas hormonas en la fisiología hace que el organismo disponga de abundantes reservas para evitar deficiencias, por ejemplo, la glándula tiroides almacena hormonas suficientes para un mes.
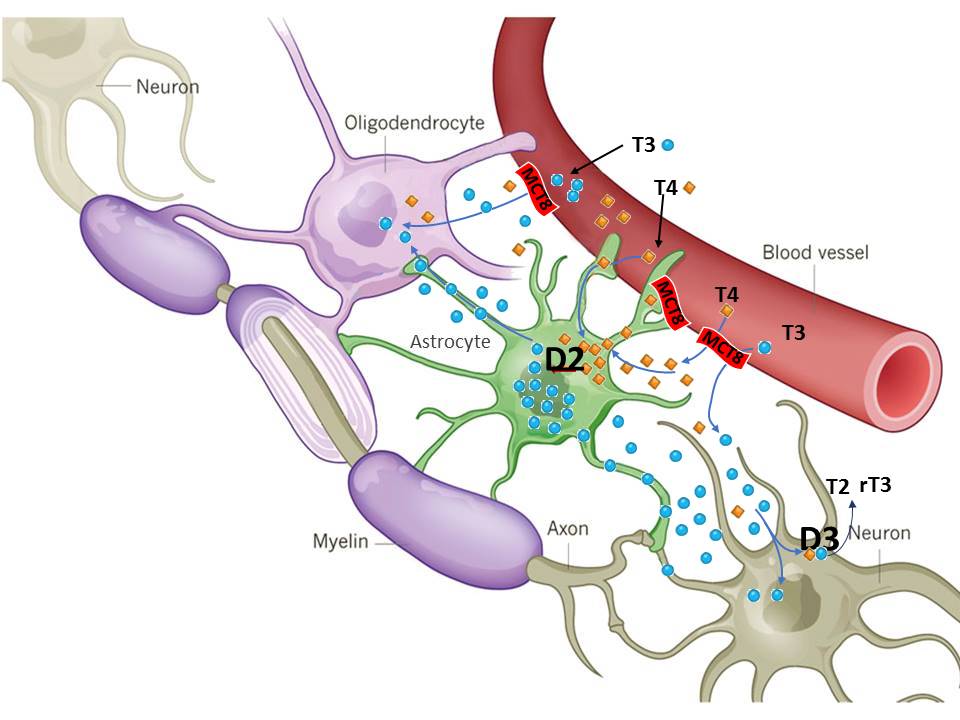
De las dos hormonas, la hormona activa es la T3. La secreción tiroidea en el ser humano consta de un 5% de T3 y un 95% de T4. Esta última es una prohormona, que se transforma en T3 en los tejidos extratiroideos mediante desyodación en 5’ catalizada por las desyodasas tipos 1 (D1) y 2 (D2), codificadas por los genes Dio1 y Dio2. La degradación de T4 y T3 se cataliza por otra desyodasa, la D3 (gen Dio3). Estas desyodasas son selenoproteínas, que en su centro activo poseen el aminoácido Se-cisteína, que usa el codón UAG que generalmente es un codón de terminación. El 80% del pool extratiroideo de T3 se produce por desyodación de la T4. Es una reacción muy importante porque las actividades D2 y D3 regulan localmente la disponibilidad de esta hormona a nivel tisular (1). La D2 se inhibe por el substrato T4, que acelera su degradación en proteasomas. En cerebro, donde la mayor parte de la T3 se origina localmente en astrocitos (2), este mecanismo mantiene las concentraciones de T3 constantes en un amplio margen de oscilación de la secreción de T4 (3).
Hace años demostramos que Dio2 se expresa en astrocitos y tanicitos, y describimos un mecanismo por el cual la T4 circulante tiene acceso a los astrocitos donde se produce la desyodación y generación de T3 (2). De los astrocitos, la T3 se transporta por mecanismos desconocidos al resto de las células neurales. La T3 actúa en las células diana regulando la expresión génica a nivel de transcripción mediante la interacción con receptores nucleares. Los receptores son factores de transcripción regulados por el ligando, que pueden activar o reprimir la expresión génica (4). Nuestro laboratorio ocupa una posición de liderazgo en la caracterización de los genes regulados por T3 en el cerebro. El número de genes bajo regulación por T3 es muy extenso. En células primarias de corteza cerebral de ratón, por ejemplo, T3 regula más de 1400 genes, y de ellos unos 300 directamente a nivel de transcripción (5).
La deficiencia de hormona tiroidea durante el desarrollo da lugar a diversos síndromes que cursan con déficit intelectual, retrasos del crecimiento y enanismo, y alteraciones neurológicas. Una causa común ha sido la deficiencia de yodo que ha afectado a gran parte de la Humanidad a lo largo de la Historia, dando lugar a bocio y cretinismo endémicos. Para evitar la deficiencia de yodo se recomienda la ingesta de sal yodada, y suplementos de yodo en embarazadas y mujeres lactantes. Otra causa es el hipotiroidismo congénito, que afecta a 1/2500 recién nacidos y es prevenible mediante el tamizado neonatal y tratamiento con T4. La eficacia del tratamiento en los casos positivos hace esta condición la causa más frecuente de deficiencia intelectual prevenible.
Pero la afección más severa es el síndrome de Allan Herdon-Dudley, causado por mutaciones en el transportador de monocarboxilatos 8 (MCT8) (6). MCT8 es una proteína de la membrana celular, de 12 dominios transmembrana, que facilita el transporte bidireccional de T3 y T4 a través de la membrana. Las mutaciones de MCT8 dan lugar a un cuadro endocrino, con retraso global del desarrollo y alteraciones de las concentraciones séricas de T4 y T3, y a un cuadro neurológico caracterizado por hipotonía neonatal severa, disquinesias paroxísticas, falta de adquisición del lenguaje y un cociente intelectual por debajo de 40. El síndrome se debe al fallo de transporte de hormona tiroidea en cerebro a través de la barrera hematoencefálica (6). Nuestro grupo, además de confeccionar una guía clínica, que propició el diagnóstico de los primeros pacientes en España, ha realizado contribuciones claves para entender la fisiopatología del síndrome, como la demostración del papel de la barrera hematoencefálica. También hemos realizado el único estudio histopatológico disponible hasta la fecha (7), que demostró la presencia de hipotiroidismo en el cerebro de feto con la mutación desde al menos la semana 30 de gestación. En la actualidad, el esfuerzo se está dirigiendo hacia la posibilidad del uso de agentes hormonales agonistas de los receptores de T3 que usen un transportador distinto de MCT8 para atravesar la barrera hematoencefálica.
Referencias:
- Hernandez A, Morte B, Belinchón MM, Ceballos A, Bernal J. (2012) Critical role oftypes 2 and 3 deiodinases in the negative regulation of gene expression by T3 in the mouse cerebral cortex. Endocrinology.153:2919-2928.
- Guadaño-Ferraz, A., M.J. Obregon, D.L. St Germain, and J. Bernal, (1997) The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci U S A,. 94:10391-10396.
- Morte B, Ceballos A, Diez D, Grijota-Martínez C, Dumitrescu AM, Di Cosmo C,Galton VA, Refetoff S, Bernal J. (2010) Thyroid hormone-regulated mouse cerebral cortex genes are differentially dependent on the source of the hormone: a study inmonocarboxylate transporter-8- and deiodinase-2-deficient mice. Endocrinology 151:2381-2387.
- Morte, B., Manzano, J., Scanlan, T., Vennström, B., & Bernal, J. (2002). Deletion of the thyroid hormone receptor alpha 1 prevents the structural alterations of the cerebellum induced by hypothyroidism. Proc Nat Acad Sci, 99: 3985–3989.
- Gil-Ibañez, P., García-García, F., Dopazo, J., Bernal, J., & Morte, B. (2017). Global Transcriptome Analysis of Primary Cerebrocortical Cells: Identification of Genes Regulated by Triiodothyronine in Specific Cell Types. Cerebral Cortex 27: 706–717.
- Bernal, J., Guadaño-Ferraz, A., & Morte, B. (2015). Thyroid hormone transporters—functions and clinical implications. Nature Rev Endocrinology, 11: 690-701.
- López-Espíndola, D., C. Morales-Bastos, C. Grijota-Martínez, X.H. Liao, D. Lev, E. Sugo, C.F. Verge, S. Refetoff, J. Bernal, and A. Guadaño-Ferraz (2014). Mutations of the thyroid hormone transporter MCT8 cause prenatal brain damage and persistent hypomyelination. J Clin Endocrinol Metab,. 99(12): p. E2799-804.