Desde su origen, el ser humano ha buscado el elixir o fuente de la eterna juventud, símbolo de la inmortalidad y la longevidad. Y gracias a las mejoras en la higiene, salud y alimentación, la esperanza de vida se ha duplicado en los últimos cien años. Sin embargo, todavía estamos muy lejos de alcanzar el tiempo que vivió la persona más longeva del planeta. Jeanne Louise Calment vivió 122 años, pero, ¿y si tuvieras que vivir toda una vida en 13 años, para luego morir de viejo?
En 1996 llegó al mundo un niño llamado Sam (1). Nació con un aspecto saludable pero pronto sus padres empezaron a ver que algo no iba bien. Sam comenzó a manifestar falta de crecimiento, pérdida de peso y cabello, arrugas y manchas en la piel, ojos saltones y rigidez. A los dos años de edad Sam fue diagnosticado con un tipo de “síndrome de envejecimiento”, el más severo, conocido como síndrome de progeria Hutchinson-Gilford. Este se caracteriza por alteraciones en la integridad del tejido conectivo -componente esencial de órganos y tejidos-, incluyendo el hueso, músculo, piel, tejido subcutáneo y vasos sanguíneos. En los próximos años desarrollaría de forma acelerada los signos característicos del envejecimiento, incluyendo problemas en las articulaciones, aterosclerosis generalizada, enfermedades cardiovasculares y derrames cerebrales. Sin embargo, mentalmente seguiría siendo un niño acorde a su edad, ya que el cerebro no se ve afectado. Su esperanza de vida era de trece años, con alta probabilidad de fallecer por enfermedad cardiovascular.
A Sam se le escapa el tiempo 7 veces más rápido que a Calment, observando con la mente de un niño envejecer su pequeño cuerpo. No se conoce ningún tratamiento ni la causa de su dolencia. Fue entonces cuando sus padres decidieron actuar. En 1999 crearon una Fundación (2) con el objetivo de encontrar un “elixir de la juventud” para niños con esta enfermedad rara que afecta a uno de cada 7 millones. Entendieron, que para curar, primero había que conocer; así que empezaron a buscar financiación para reclutar y apoyar a los científicos en su labor de encontrar la causa y un tratamiento para niños como Sam.
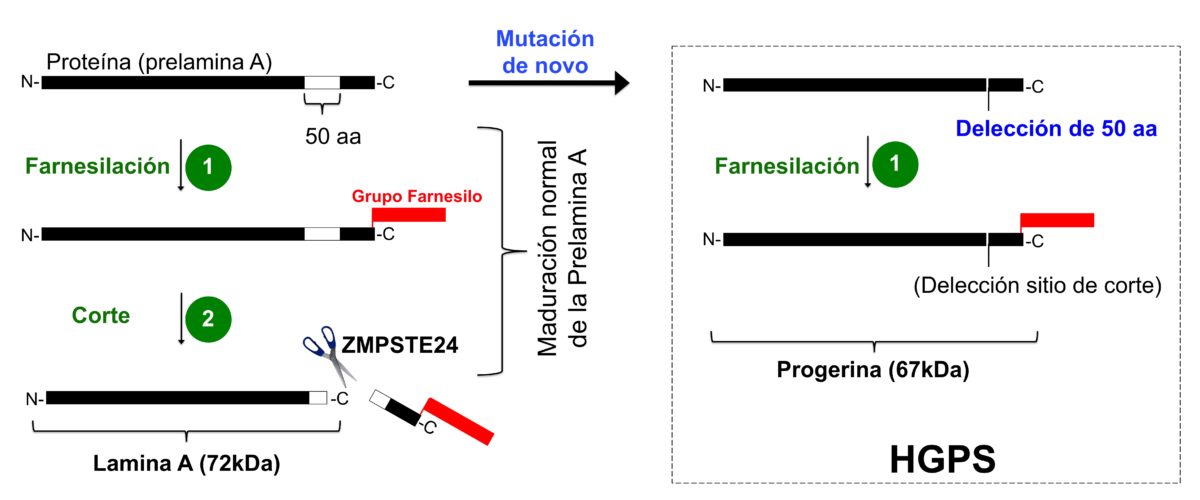
De esta forma, en 2003 se encuentra la causa molecular de la enfermedad: la presencia de una proteína anómala llamada progerina. Esta proteína se produce como consecuencia de una mutación nueva que aparece por primera vez en una familia (se denomina “de novo”). La mutación es poco frecuente y aparece en el zigoto o en una célula germinal de los padres (espermatozoides y óvulos).
La mutación se produce en el gen de la lámina A (ver figura), proteína relacionada con la estructura del núcleo de la célula. Esta mutación produce una proteína aberrante con 50 aminoácidos menos, que es esencial en el proceso normal de maduración de la lámina A. Esta versión corta de la lámina A es conocida como progerina y lleva además una molécula hidrofóbica por farnesilación, que la hace más tóxica, generando aberraciones en la estructura nuclear y en la expresión de genes desde el núcleo. Sam produce progerina debido a su mutación; sus padres, al igual que muchas personas, probablemente la producirán y acumularán al envejecer.
Este descubrimiento constituye un punto de inflexión muy importante en la lucha contra esta enfermedad. Por un lado, su diagnóstico es posible y permite informar con detalle a los padres cuyos hijos nacen como Sam. Por otro lado, los científicos pueden estudiar los efectos de la progerina en tejidos y órganos. Para este fin, la creación de un ratón transgénico con la misma mutación genética que Sam, puede ser clave en la lucha contra esta enfermedad que padecen menos de ciento cincuenta niños en todo el mundo. Los ratones que producen progerina, generan las mismas manifestaciones clínicas que Sam. Gracias a estos ratones, investigadores de todo el mundo han encontrado varios mecanismos moleculares que explican no solamente el envejecimiento acelerado, sino también otros mecanismos relacionados con el envejecimiento normal. Ello está permitiendo el desarrollo experimental de varios tratamientos, incluyendo terapias que evitan la farnesilación, es decir la inclusión de la molécula hidrofóbica que lleva la lámina A aberrante. Desafortunadamente, su aplicación en niños es muy lenta y costosa, y solo permiten mejorar los síntomas principales (3).
El tratamiento definitivo consistirá en la eliminación de la mutación genética. Todavía falta tiempo para lograr esto de forma efectiva y segura en los humanos. Sin embargo, científicos españoles han dado un pequeño paso para lograrlo (4). Utilizando la técnica de edición genética CRISPR (5), estos científicos han logrado revertir dicha mutación en un pequeño porcentaje de células; logrando, con ello, alargar hasta un 25% la vida del ratón transgénico progeroide. No obstante, todavía estamos lejos de poder aplicarlo en niños con progeria.
Se estima que en 2040 España será el país con la mayor esperanza de vida de todo el planeta (85,8 años). Sam murió a los 17 años de edad disfrutando de más tiempo del esperado y sabiendo que no hay que desaprovechar el tiempo, esté acortado o no.
REFERENCIAS
- https://www.youtube.com/watch?v=36m1o-tM05g
- https://www.progeriaresearch.org
- Gordon LB, et al. Clinical trial of a farnesyl transferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012 Oct 9;109(41):16666-71. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3478615/pdf/pnas.201202529.pdf
- https://www.elespanol.com/ciencia/salud/20190219/golpe-espanol-progeria-enfermedad-convierte-ninos-ancianos/377213050_0.html
- https://web2020.sebbm.es/web/es/divulgacion/rincon-profesor-ciencias/articulos-divulgacion-cientifica/2159-editando-genomas-con-las-herramientas-crispr